Path to LALD Diagnosis
Our service is ideal for identification of LAL Deficiency in at-risk patients showing specific clinical symptoms or for an individual or family member who has a family history of LALD.
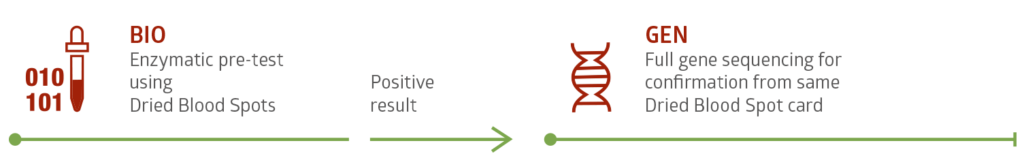
Testing is fast and safe using Dried Blood Spot (DBS) cards. This simple and minimally invasive technique supplies enough sample for biochemical testing and in most cases genetic confirmation testing as well.
Available enzymatic and genetic tests:
| Disease | Enzyme Tests | Biomarker Tests | Genetic Tests |
|---|---|---|---|
| Lysosomal acid Lipase deficiency | LAL (Lysosomal acid Lipase) | -- | LIPA |
Differential diagnosis options:
| Disease | Enzyme Tests | Biomarker Tests | Genetic Tests |
|---|---|---|---|
| Gaucher Disease | GBA (acid β-glucosidase) | Lyso-GL-1 (Lyso-Gb1) | GBA |
| ASMD (Niemann-Pick Type A/B) | ASM (Acid Sphingomyelinase) | Lyso-Sphingomyelin (Lyso-SPM) | SMPD1 |
| Niemann-Pick Type C1/D | -- | -- | NPC1 |
| Niemann-Pick Type C2 | -- | -- | NPC2 |
Quality:
Fully validated and accredited* according to the highest quality standards for Medical Laboratories (ISO 15189).
Methodologies:
- Enzyme assays by Clinical Mass Spectrometry.
- Genetics by Sanger and Next-Generation Sequencing platforms.
About LALD
What is LAL deficiency?
Lysosomal acid Lipase deficiency (LALD) is an autosomal recessive genetic disease. Lysosomal acid Lipase (LAL) plays an important role in breaking down cholesteryl esters and triglycerides into low density lipoprotein particles, free cholesterol, and free fatty acids. The lack of the LAL enzyme can lead to cholesteryl esters and triglycerides accumulating in a number of body organs including the liver, spleen, gut, blood vessel walls, and other important organs.
Which mutation causes an enzyme deficiency?
The condition is caused by a mutation of the LIPA gene, which is responsible for the gene coding of the lysosomal Lipase protein (also called lysosomal acid Lipase or LAL), which results in a loss of normal protein function.
Are there diseases similar symptoms to LAL deficiency?
Clinical presentation of LAL deficiency is similar to other congentital sphingolipidoses which can result in underdiagnosis. Infants, children, and adults that suffer from LALD experience a range of serious health problems. The accumulation of fat in the walls of the gut and other organs in leads to serious digestive problems including malabsorption, a condition in which the gut fails to absorb nutrients and calories from food.
How to Order LALD Diagnostic Services

Our Diagnostic Service for LAL deficiency includes enzyme testing for Lysosomal acid lipase as well as any necessary genetic molecular analysis.
All of our services are available to any interested physician or healthcare professional worldwide.
As part of our diagnostic services, we supply complimentary ARCHIMEDlife sampling kits. You can order your sampling kits and diagnostic services through our easy and secure WEBPORTAL and receive your electronic medical report in five simple steps.
Five Simple Steps

1Order Sampling Kit

2Collect the Sample

3Register the DBS Card

4Return the Sample

5Receive your Report