Path to GM1 and GM2 Gangliosidosis Diagnosis
Our service is an excellent option for the identification of GM1 and GM2 Gangliosidosis in at-risk patients showing specific clinical symptoms or for an individual or family member who has a family history of Gangliosidosis.
Testing is fast and safe using Dried Blood Spot (DBS) cards. This simple and minimally invasive technique supplies enough samples for biochemical testing and in most cases genetic confirmation testing as well.
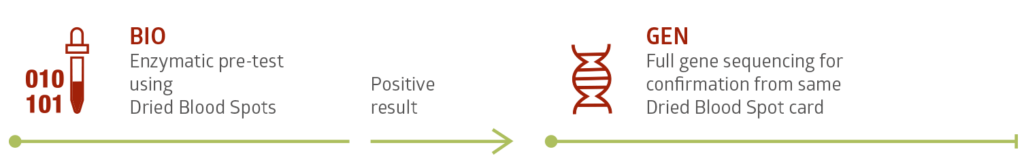
Available enzymatic and genetic tests:
| Disease | Enzyme Tests | Genetic Tests |
|---|---|---|
| GM1 Gangliosidosis (GM1 Type I-III, MPS IVB) | β-Galactosidase | GLB1 |
| Tay–Sachs disease | β-Hexosaminidase A | HEXA |
| Sandhoff disease | β-Hexosaminidase A and B | HEXB |
| GM2-gangliosidosis, AB variant | -- | GM2A |
Differential diagnosis options:
| Disease | Enzyme Tests | Genetic Tests |
|---|---|---|
| Mucopolysaccharidoses (MPSs) | Multiple enzymes | ARSB, GALNS, GLB1, GNS, GUSB, HGSNAT, HYAL1, IDS, IDUA, NAGLU, SGSH |
| Mucolipidosis I | -- | NEU1 |
| Mucolipidosis II alpha/beta Mucolipidosis III alpha/beta | -- | GNPTAB |
| Mucolipidosis III gamma | -- | GNPTG |
| Mucolipidosis IV | -- | MCOLN1 |
Quality:
Fully validated and accredited* according to the highest quality standards for Medical Laboratories (ISO 15189).
Methodologies:
- Enzyme assays by Clinical Mass Spectrometry.
- Genetics by Sanger and Next-Generation Sequencing platforms.
About GM1 and GM2 Gangliosidosis
Gangliosidoses are a group of Lysosomal Storage Disorders that progressively destroy nerve cells (neurons) in the brain and spinal cord.
GM1 Gangliosidosis
Classified into three major types based on the age of disease onset:
- Classic infantile (Type I, up to 6 months of age)
- Juvenile (Type II, 18 months to 5 years of age)
- Adult onset/chronic (Type III, > 5 years of age)
GM2 Gangliosidosis
Group of three related genetic disorders:
- Tay–Sachs disease
- GM2 Gangliosidosis, Variant AB
- Sandhoff disease
Which mutation causes an enzyme deficiency?
These disorders are caused by a genetic mutation resulting in different enzyme deficiencies. Deficient enzyme activity leads to toxic accumulation of gangliosides in body tissues, particularly in the central nervous system (CNS). The degree of accumulation of gangliosides in visceral organs, including the liver, is variable and depends on the disease subtype. Disorders related to defects of ganglioside synthesis are very rare.
Are there diseases with similar symptoms to GM1 or GM2 gangliosidosis?
Common symptoms such as corneal clouding and hepatosplenomegaly are seen in other lysosomal storage disorders such as Mucolipidosis Type I and Mucopolysaccharidoses Types I and II. Developmental delay and growth abnormalities are also observed with Mucolipidosis Types II and III.
How to Order GM1 and GM2 Gangliosidosis Diagnostic Services

Our Diagnostic Service for GM1 and GM2 Gangliosidosis includes enzyme testing for β-Galactosidase (GM1 Gangliosidosis), β-Hexosaminidase A (Tay–Sachs disease) and β-Hexosaminidase A and B (Sandhoff disease) as well as any necessary genetic molecular analysis.
All of our services are available to any interested physician or healthcare professional worldwide.
As part of our diagnostic services, we supply complimentary ARCHIMEDlife sampling kits. You can order your sampling kits and diagnostic services through our easy and secure WEBPORTAL and receive your electronic medical report in five simple steps.
Five Simple Steps

1Order Sampling Kit

2Collect the Sample

3Register the DBS Card

4Return the Sample

5Receive your Report