Mucopolysaccharidoses (MPSs) are a group of chronic, progressive lysosomal storage diseases (LSDs) with multi-system impairments. They are rare genetic disorders with a combined incidence of approximately 1 in 25,000 live births (Tomatsu S et al 2013) and the most common type is MPS II followed by MPS I, III, IV, VI and VII (Khan SA et al 2017). These disorders are caused by an enzyme deficiency resulting in the inability to break down glycosaminoglycans (GAGs; formerly called mucopolysaccharides) into smaller sugar molecules. Glycosaminoglycans in lysosomes include chondroitin sulfate, dermatan sulfate, heparin sulfate, keratin sulfate and/or hyaluronan. An enzyme deficiency leads to an accumulation of GAGs in arteries, eyes, skeleton, joints, skin, ears and/or teeth. Glycosaminoglycans can also accumulate in the respiratory system, spleen, liver, central nervous system, bone marrow and blood (rarediseases.org). Depending on which of the eleven known enzymes are affected as well as the level of enzyme activity, different clinical manifestations are described varying from mild to severe forms with potentially early death. At present, MPSs are summarized in seven syndrome types (see Table 1 below) (orphanet 2018).
Mucopolysaccharidosis (MPS)
Table1: Different syndromes of MPS.
| Type | OMIM# | Name | Enzyme | Gene |
| MPS I | 607014 607015 |
Hurler (IH) Hurler-Scheie (IH/S) |
alpha-L-iduronidase | IDUA |
| MPS II | 309900 | Hunter | Iduronate-2-Sulfatase | IDS |
| MPS IIIA MPS IIIB MPS IIIC MPS IIID |
252900 252920 252930 252940 |
Sanfilippo A Sanfilippo B Sanfilippo C Sanfilippo D |
Heparan N-sulfatase (sulfamidase) N-acetyl-alpha-glucosaminidase acetyl CoA alpha-glucosaminide acetyltransferase N-acetylglucosamine-6-sulfatase |
SGSH NAGLU HGSNAT GNS |
| MPS IVA MPS IVB |
253000 253010 |
Morquio A Morquio B |
N-acetyl-galactosamine-6-sulfate sulfatase beta-galactosidase |
GALNS GLB1 |
| MPS V | – | – | – | – |
| MPS VI | 253200 | Maroteaux-Lamy | arylsulfatase B (N-acetylgalactosamine 4-sulphatase) |
ARSB |
| MPS VII | 253220 | Sly | beta-glucuronidase | GUSB |
| MPS VIII | – | – | – | – |
| MPS IX | 601492 | Natowicz | hyaluronidase | HYAL1 |
Inheritance
Inheritance is mostly autosomal recessive and with MPS II X-linked recessive. In the situation of an autosomal recessive inheritance with both parents carrying one faulty gene, 25% of their offspring will be healthy, 25% will be affected by MPS and half of their offspring will be carriers (Figure 1). In the case of a X-linked recessive inheritance, the faulty gene is on the X chromosome. A defective X chromosome in females is deactivated. Consequently, these women will not show clinical symptoms, however they are carriers for MPS. Women who are genetic carriers for X-linked recessive MPS II have a 25% chance to have a carrier daughter, a 25% of having an MPS affected son and a 50% chance of an unaffected son or daughter (Figure 2a and 2b). Males inheriting the faulty gene, will be affected with MPS. In the next generation, daughters of affected MPS fathers will be carriers.
Figure 1: Autosomal recessive inheritance of MPS I, IIIB, IVA, VI and VII.
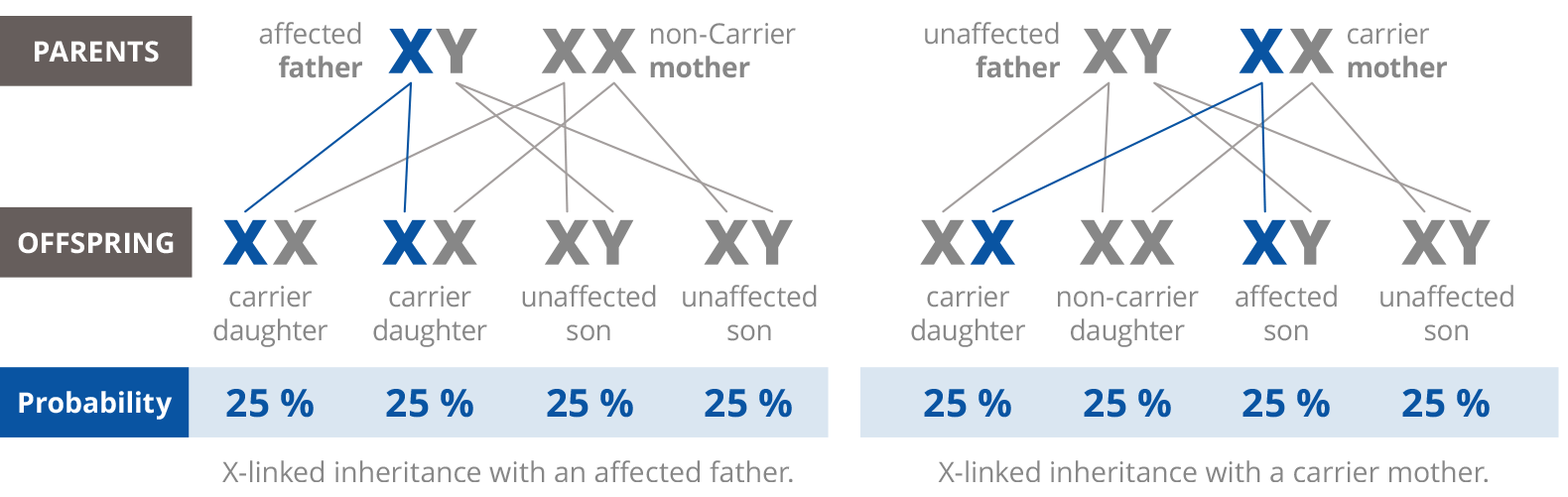
Figure 2a and 2b: X-linked recessive inheritance of MPS II.
Clinical manifestations of MPS
- Prenatal abnormalities: Clinical manifestations prenatally include none-immune fetal hydrops and spontaneous abortion (Venkat-Raman N et al 2006, Vianey-Saban C et al 2016, Kubaski F et al 2017) caused by MPS.
- Severe forms of MPS: Affected newborns usually are “healthy”. Initially, affected infants develop recurrent infections (urinary system, ear or respiratory tract), growth retardation and mild neurodevelopmental delay during the first years of life with progression. Clinical manifestations comprise atypical facial features, corneal clouding and retinal degeneration, hearing loss, systemic impairment of skeleton including joints, short stature and growth impairment, heart and lung problems, hepatosplenomegaly, neurological involvements, seizures, progressive dementia, aggressive behavior, intellectual disability and possible early death. The severity of MPS varies in affected people even inside the family. Due to the fact that MPS is a progressive disorder, clinical course can be influenced through specific treatment. Therefore, an early diagnosis and treatment before clinical manifestation is very important (Melbouci M et al 2018, Nagao K et al 2018, mpssociety.org.uk, rarediseases.org)
- Mild forms of MPS: These individuals may become clinically symptomatic during childhood or even adolescence.
Promising treatment options
Today, different specific treatment strategies are available for affected infants with MPS like enzyme replacement therapy (ERT), substrate reduction therapy (SRT) or transplantation of hematopoietic stem cells (HSCT) with long-term benefits (mpssociety.org.uk). At present, it is not possible to “cure” MPS, although it is possible to influence the clinical course of MPS, reducing the complication rate. Therefore, adequate treatment improves life quality of affected infants and their families. A novel promising treatment strategy is the neonatal cellular and gene therapy (Tomatsu S et al 2016, Sawamoto K et al 2018).
Important information for using the Dried Blood Spot card.
Apply only one blood drop in each circle of the filter paper card (DBS card) to one side. Dry the card with free air flow at room temperature at least 4 hours before you put the card into the envelope for transportation. Please return the card within 3 days. In case you cannot send the DBS card immediately and you have to store the sample longer or for any other information, please contact us: info@archimedlife.com.
You will find further detailed information about diagnostic testing using the Dried Blood Spot card online:
www.archimedlife.com/dried-blood-spot.
References:
- Khan SA, Peracha H, Ballhausen D, Wiesbauer A, Rohrbach M, Gautschi M, Mason RW, Giugliani R, Suzuki Y, Orii KE, Orii T, Tomatsu S. Epidemiology of mucopolysaccharidoses. Mol Genet Metab. 2017;121:227-240.
- Kubaski F, Brusius-Facchin AC, Mason RW, Patel P, Burin MG, Michelin-Tirelli K, Kessler RG, Bender F, Leistner-Segal S, Moreno CA, Cavalcanti D, Giugliani R, Tomatsu S. Elevation of glycosaminoglycans in the amniotic fluid of a fetus with mucopolysaccharidosis VII. Prenat Diagn. 2017;37:435-439.
- Melbouci M, Mason RW, Suzuki Y, Fukao T, Orii T, Tomatsu S. Growth impairment in mucopolysaccharidoses. Mol Genet Metab. 2018: S1096-7192(18)30147-1.
- mpssociety.org.uk
- Nagao K, Morlet T, Haley E, Padilla J, Nemith J, Mason RW, Tomatsu S. Neurophysiology of hearing in patients with mucopolysaccharidosis type IV. Mol Genet Metab. 2018:S1096-7192(18)30075-1.
- Rarediseases.org. NORD National Organization for Rare Disorder
- Sawamoto K, Chen HH, Alméciga-Díaz CJ, Mason RW, Tomatsu S. Gene therapy for Mucopolysaccharidoses. Mol Genet Metab. 2018 Feb;123(2):59-68.
- Tomatsu S, Azario I, Sawamoto K, Pievani AS, Biondi A, Serafini M. Neonatal cellular and gene therapies for mucopolysaccharidoses: the earlier the better? J Inherit Metab Dis. 2016;39:189-202.
- Tomatsu S, Fujii T, Fukushi M, Oguma T, Shimada T, Maeda M, Kida K, Shibata Y, Futatsumori H, Montaño AM, Mason
RW, Yamaguchi S, Suzuki Y, Orii T. Newborn screening and diagnosis of mucopolysaccharidoses. Mol Genet Metab. 2013;110:42-53. - Venkat-Raman N, Sebire NJ, Murphy KW. Recurrent fetal hydrops due to mucopolysaccharidoses type VII. Fetal Diagn Ther. 2006;21:250-4.
- Vianey-Saban C, Acquaviva C, Cheillan D, Collardeau-Frachon S, Guibaud L, Pagan C, Pettazzoni M, Piraud M, Lamazière A, Froissart R. Antenatal manifestations of inborn errors of metabolism: biological diagnosis. J Inherit Metab Dis. 2016;39:611-624.