This data was presented during the 11 February 2021 poster session of the 17th WORLDSymposium (virtual).
Abstract
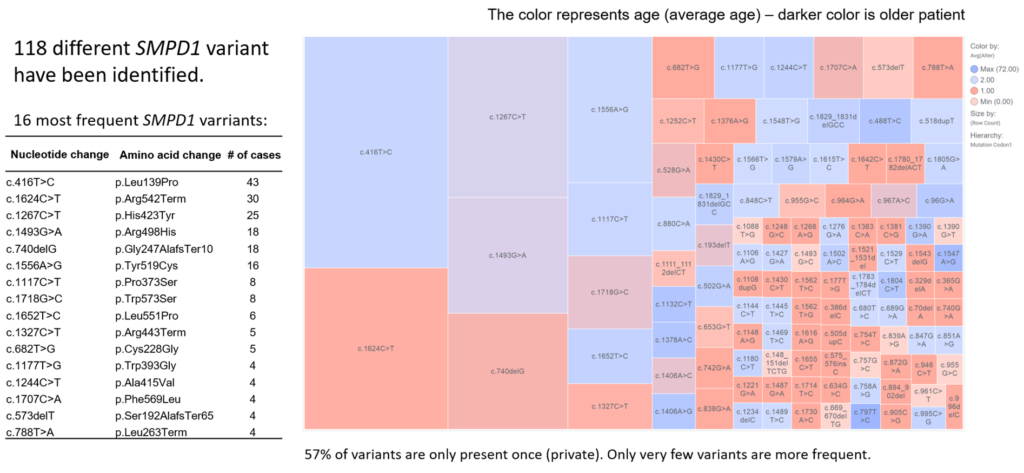
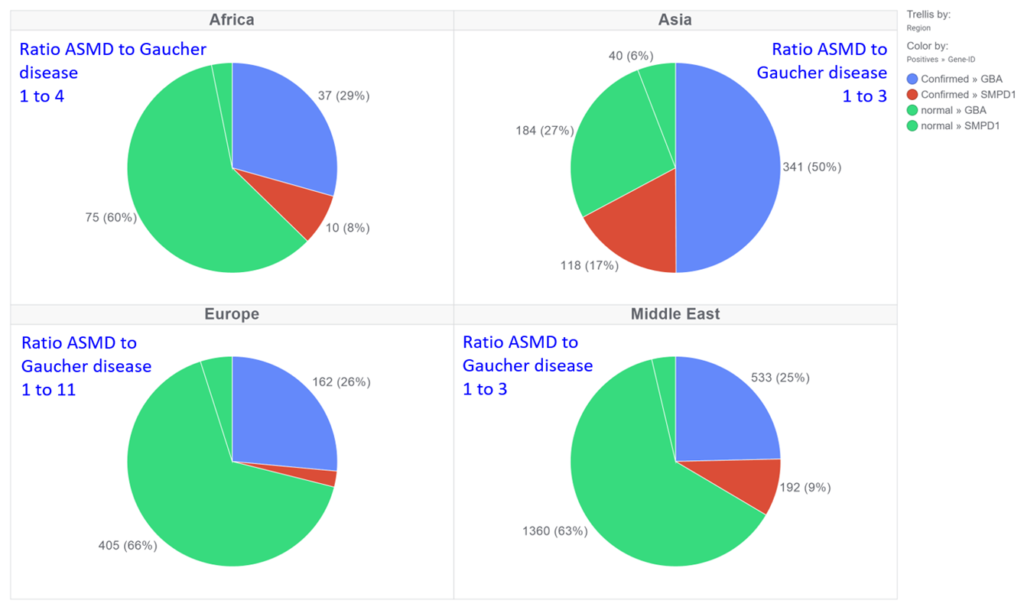
In this prospective study, samples from 14,830 individuals from 61 countries were tested using a combined biochemical and genetic approach. For all samples, β-Glucocerebrosidase (GBA) and acid sphingomyelinase (ASM) enzyme activities, were measured simultaneously in dried blood spot specimens (DBS) by tandem mass spectrometry. Deficient enzyme activity was detected in 3,585 DBS (3097 were GBA-deficient and 488 were ASM-deficient). Genotype analysis of GBA or SMPD1 resulted in 1073 confirmed diagnoses of Gaucher disease and 335 confirmed diagnoses of ASMD. The ratio of ASMD to Gaucher disease patients diagnosed varied by region and country. The highest in ratios were seen in Asia and Middle Eastern countries (1 to 3), followed by Africa (1 to 4) and Europe (1 to 11). In the Middle East and Asia, the highest ratio of ASMD to Gaucher disease patients diagnosed was in Iraq where 54 patients were diagnosed with ASMD and 50 with Gaucher disease. Iran, and Pakistan, and Turkey similarly high ratios. In Europe, the highest ratio of ASMD to Gaucher disease patients diagnosed was in Romania where five patients were diagnosed with ASMD and seven with Gaucher disease. The lowest ratio in Europe was in Italy where one patient was diagnosed with ASMD and ten with Gaucher disease. Most of the ASMD patients were newborns and children under 10 years of age. 110 distinct SMPD1 sequence variants were identified. Most of the variants are present once. Ten variants were more frequent (p.Leu139Pro; p.Arg542Term; p.His423Tyr; p.Arg498His; p.Gly247AlafsTer10; p.Tyr519Cys; p.Pro373Ser, p.Trp573Ser, p.Leu551Pro; p.Arg443Term; p.Cys228Gly). Here we would like to further present our genotype-phenotype correlation together with enzyme activity and Lyso-SPM biomarker levels.
Introduction
Gaucher (GBA deficiency) and Niemann-Pick A/B disease (acid sphingomyelinase (ASM) deficiency)) are autosomal recessive inherited disorders of metabolism that result from a deficiency of the enzymes glucocerebrosidase and acid-sphingomyelinase, respectively. Due to patients with Gaucher and Niemann-Pick A/B disease presenting with similar and overlapping clinical symptoms, a systematic laboratory workup evaluating both diseases in parallel is very important.
The overall estimated incidence is 0.4 to 0.6 in 100,000 live births, and although ASMD is rare, this may be an underestimate of the true incidence due to under- or mis-diagnosis. ASMD represents a wide clinical spectrum of disease with varying symptoms at presentation, age of onset, and degree and type of organ and systemic involvement. The disease manifestations can vary, but frequently involve hepatosplenomegaly with progressive organ dysfunction, interstitial lung disease, and bleeding. The cellular damage caused by sphingomyelin accumulation can be irreversible and can lead to life-threatening complications and reduced life expectancy. Quality of life is an important consideration for patients with ASMD and their families, as the disease has a substantial impact on functioning and day-to-day living.
A lack of disease awareness and non-specific presentation can result in delayed diagnosis. Several diseases (including primary hepatic disease, Gaucher disease, NPD C, and lysosomal acid lipase deficiency) present with symptoms similar to ASMD, and comparative assessments may be useful for a differential diagnosis.
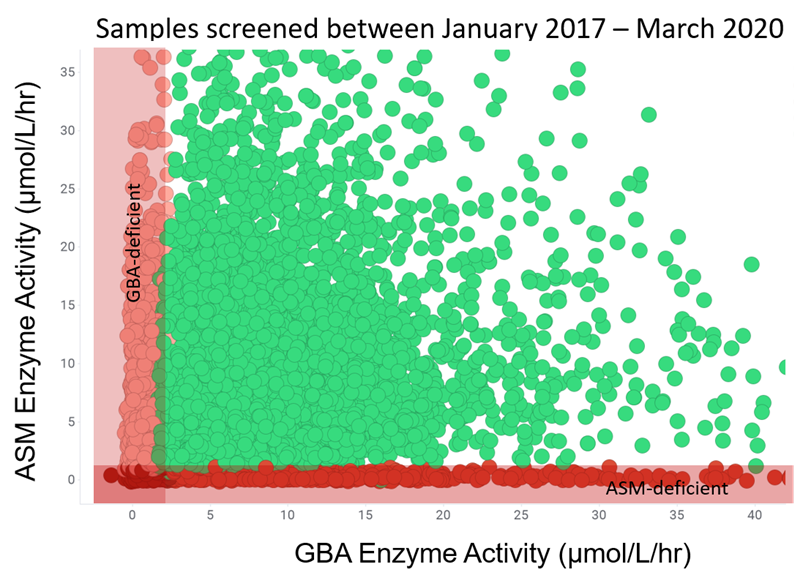
Enzyme activity screening
Deficient enzyme was detected in 3,585 DBS samples:
samples screened 14,830
GBA-deficient 3,097 (21%)
ASM-deficient 488 (3%)
Samples in the study are based on high-risk population screening. ASMD positive cases are represented by the red dots.
Genetic Confirmatory Testing
488 cases have been submitted to genetic analysis.
Number of cases with SPMD1 variant
Two 335 (69%)
One 29
No 124
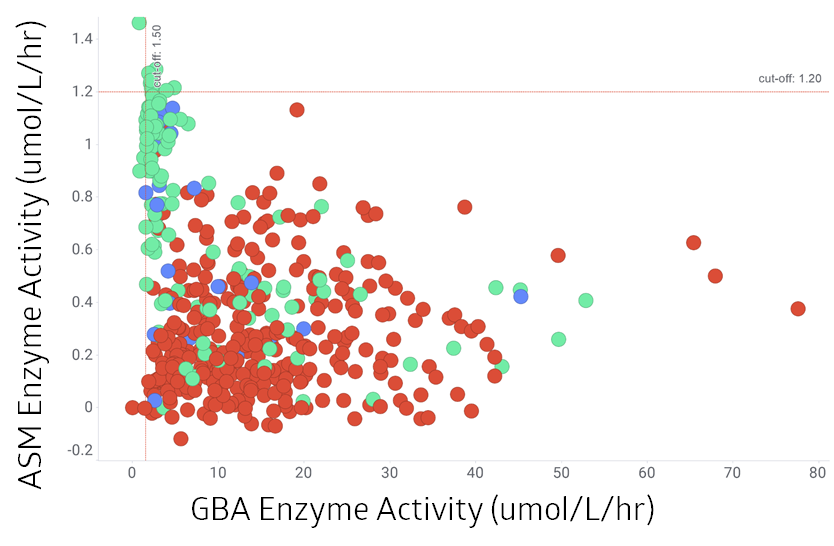
Table of Variants
ASMD (335) to Gaucher Disease (1073)
Ratio SPM 509 MoM*/Lyso-SPM
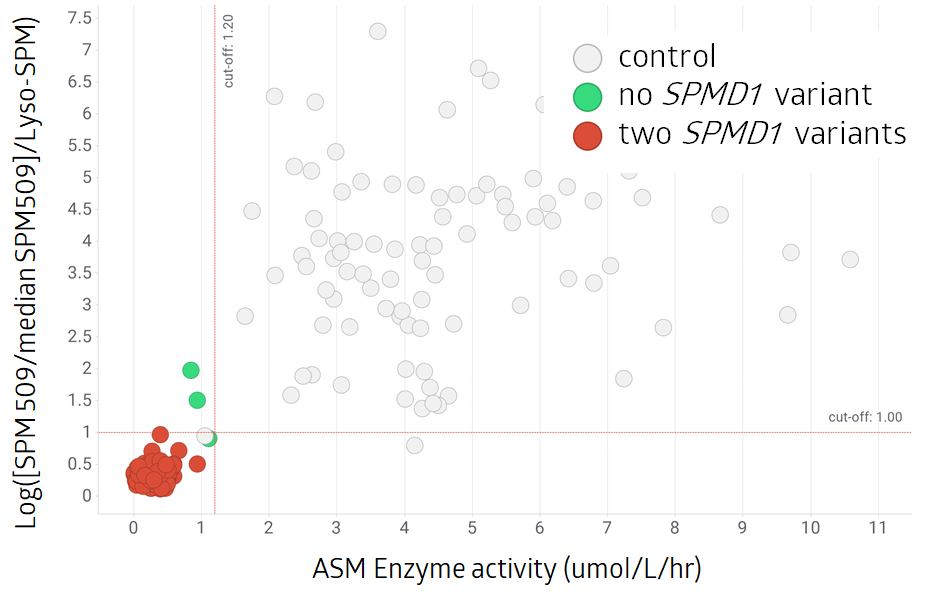
In total: 239 cases
Number of cases with SMPD1 variants:
Two 84
No 3
152 control samples
By using the ratio of SPM 5091 and Lyso-SPM as previously published1 the cases with no SPMD1 variant can be further separated from those having two SPMD1 variants. Control samples are clearly separated from the other two groups.
(2 dots are not shown on the graph with ASM activity 14.62 umol/L/hr and 18.16 umol/L/hr.)
*multiple of the median (MoM)
Conclusions
Our results have shown that on average 1 out of 5 suspected Gaucher patients is suffering from ASM deficiency resulting in Niemann-Pick A/B disease diagnosis (frequency depends on region). For this reason, it is recommended to test suspected patients for both Gaucher and Niemann-Pick diseases simultaneously.
In addition, Lyso-SPM shows promise as a useful biomarker for ASMD patients for both diagnostics and monitoring. Further prospective studies are needed to provide more evidence.
References
- Deodato F, Boenzi S, Taurisano R, et al. The impact of biomarkers analysis in the diagnosis of Niemann-Pick C disease and acid sphingomyelinase deficiency. Clin Chim Acta. 2018;486:387–394. doi:10.1016/j.cca.2018.08.039
Authors
Petra Oliva1, Markus Schwarz1, Jake Scott1, Stefaan Sansen2, Thomas P. Mechtler1, Joan Keutzer3, Berthold Streubel4, Edward H. Schuchman5, David C. Kasper1
- ARCHIMED Life Science GmbH, Vienna, Austria
- Sanofi Genzyme, Amsterdam, Netherlands
- Cambridge, MA, USA
- The Medical University of Vienna, Department of Pathology, Vienna, Austria
- Icahn School of Medicine at Mount Sinai, New York, NY, USA