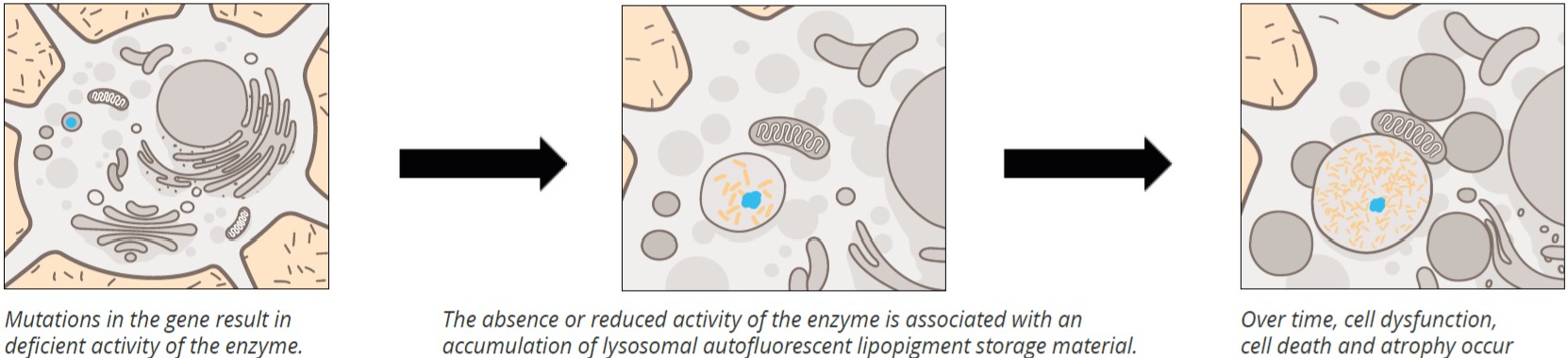
The Neuronal Ceroid Lipofuscinoses (CLN) are a group of inherited neurodegenerative disorders that affect children and adults. They are grouped together by similar clinical features and the accumulation of auto-fluorescent storage material. More than a dozen genes containing over 430 mutations have been identified to cause at least 13 known types of NCLs (Table 1).
The clinical differential diagnosis of the NCL types is based on age of onset, clinical phenotype, ultra structural characterization of the storage material and enzyme levels. Symptoms associated with these disorders can vary widely. Although protein dysfunction or lipopigment accumulation influences many cells, brain cells are typically affected first.
CLN1 disease (classic infantile) affecting infants, with an onset of symptoms between 6 and 24 months of age.
CLN2 disease (late-infantile) affects children between the ages of 2 and 4. Consequently, an adequate and differential diagnosis is the highest priority.